Thalassemia....mmmm adik iparku dua orang ada penyakit nie...alhamdulilah suamiku tidak pembawa gen Thalassemia ....awal perkahwinan risau jgk yelah saya dah pregnant 2 bulan lbh rasanya baru kami berdua check darah nak pastikan kami berdua tidak pembawa tersebut....kami berdua bertawakkal jer...yelah saya dah pregnant...riasu jgk....alhamdulilah keputusan darah tiada. Syukur sangat2.....sekarang di Terengganu pasangan yg nak berkahwin kena check darah nak memastikan tiada pembawa....info kat bawah nie pun cari kat google jer.....
Apakah Thalassemia itu?
Thalassemia adalah penyakit keturunan dengan gejala utama pucat, perut tampak membesar karena pembengkakan limpa dan hati, apabila tidak diobati dengan baik akan terjadi perubahan bentuk tulang muka dan warna kulit menjadi menghitam. Penyebab penyakit ini adalah kekurangan salah satu zat pembentuk hemoglobin (Hb) sehingga produksi hemoglobin berkurang.
Apakah hemoglobin?
Hemoglobin adalah suatu zat di dalam sel darah merah yang berfungsi mengangkut zat asam dari paru-paru ke seluruh tubuh, selain itu yang memberikan warna merah sel darah merah. Hemoglobin terdiri dari 4 molekul zat besi (heme), 2 molekul rantai globin alpha dan 2 molekul rantai globin beta. Rantai globin alpha dan beta adalah protein yang produksinya disandi oleh gen globin alpha dan beta.
Apakah gen globin alpha dan gen globin beta?
Setiap sifat dan fungsi fisik pada tubuh kita dikontrol oleh gen, yang bekerja sejak masa embrio. Gen terdapat di dalam setiap sel tubuh kita. Setiap gen selalu berpasangan. Satu belah gen berasal dari ibu, dan yang lainnya dari ayah. Diantara banyak gen dalam tubuh kita, terdapat sepasang gen yang mengontrol pembentukan hemoglobin pada setiap sel darah merah. Gen tersebut dinamakan gen globin. Gen-gen tersebut terdapat di dalam kromosom.
Bagaimana terjadinya penyakit thalassemia?
Penyakit thalassemia disebabkan oleh adanya kelainan/perubahan/mutasi pada gen globin alpha atau gen globin beta sehingga produksi rantai globin tersebut berkurang atau tidak ada. Akibatnya produksi Hb berkurang dan sel darah merah mudah sekali rusak atau umurnya lebih pendek dari sel darah normal (120 hari). Bila kelainan pada gen globin alpha maka penyakitnya disebut thalassemia alpha, sedangkan kelainan pada gen globin beta akan menyebabkan penyakit thalassemia beta. Karena di Indonesia thalassemia beta lebih sering didapat maka selanjutnya kami hanya akan menjelaskan mengenai thalassemia beta.
Bagaimana cara penurunannya?
Penyakit ini diturunkan melalui gen yang disebut sebagai gen globin beta yang terletak pada kromosom 11. Pada manusia kromosom selalu ditemukan berpasangan. Gen globin beta ini yang mengatur pembentukan salah satu komponen pembentuk hemoglobin. Bila hanya sebelah gen globin beta yang mengalami kelainan disebut pembawa sifat thalassemia-beta. Seorang pembawa sifat thalassemia tampak normal/sehat, sebab masih mempunyai 1 belah gen dalam keadaan normal (dapat berfungsi dengan baik). Seorang pembawa sifat thalassemia jarang memerlukan pengobatan. Bila kelainan gen globin terjadi pada kedua kromosom, dinamakan penderita thalassemia (Homosigot/Mayor). Kedua belah gen yang sakit tersebut berasal dari kedua orang tua yang masing-masing membawa sifat thalassemia.
Pada proses pembuahan, anak hanya mendapat sebelah gen globin beta dari ibunya dan sebelah lagi dari ayahnya. Bila kedua orang tuanya masing-masing pembawa sifat thalassemia maka pada setiap pembuahan akan terdapat beberapa kemungkinan. Kemungkinan pertama si anak mendapatkan gen globin beta yang berubah (gen thalassemia) dari bapak dan ibunya maka anak akan menderita thalassemia. Sedangkan bila anak hanya mendapat sebelah gen thalassemia dari ibu atau ayah maka anak hanya membawa penyakit ini. Kemungkinan lain adalah anak mendapatkan gen globin beta normal dari kedua orang tuanya.

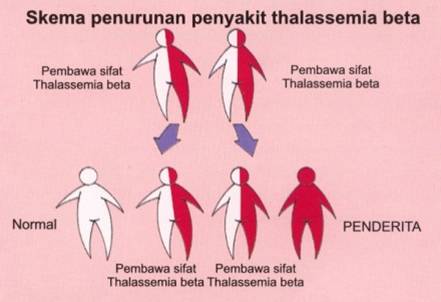
Dari skema diatas dapat dilihat bahwa kemungkinan anak dari pasangan pembawa sifat thalassemia beta adalah 25% normal, 50% pembawa sifat thalassemia beta, dan 25% thalassemia beta mayor (anemia berat).
Bagaimana terjadinya gejala pucat atau anemia?
Warna merah dari darah manusia disebabkan oleh hemoglobin yang terdapat di dalam darah merah. Hemoglobin terdiri atas zat besi dan protein yang dibentuk oleh rantai globin alfa dan rantai globin beta. Pada penderita thalassemia beta, produksi rantai globin beta tidak ada atau berkurang. Sehingga hemoglobin yang dibentuk berkurang. Selain itu berkurangnya produksi rantai globin beta mengakibatkan rantai globin alfa relatif berlebihan dan akan saling mengikat membentuk suatu benda yang menyebabkan sel darah merah mudah rusak. Berkurangnya produksi hemoglobin dan mudah rusaknya sel darah merah mengakibatkan penderita menjadi pucat atau anemia atau kadar Hbnya rendah.
Mengapa limpa membesar pada penderita thalassemia?
Limpa berfungsi membersihkan sel darah yang sudah rusak. Selain itu limpa juga berfungsi membentuk sel darah pada masa janin. Pada penderita thalassemia, sel darah merah yang rusak sangat berlebihan sehingga kerja limpa sangat berat. Akibatnya limpa menjadi membengkak. Selain itu tugas limpa lebih diperberat untuk memproduksi sel darah merah lebih banyak.
Mengapa terjadi perubahan bentuk tulang muka?
Sumsum tulang pipih adalah tempat memproduksi sel darah. Tulang muka adalah salah satu tulang pipih, Pada thalassemia karena tubuh selalu kekurangan darah, maka pabrik sel darah daiam hal ini sumsum tulang pipih akan berusaha memproduksi sel darah merah sebanyak-banyaknya. Karena pekerjaannya yang meningkat maka sumsum tulang ini akan membesar, pada tulang muka pembesaran ini dapat dilihat dengan jelas dengan adanya penonjolan dahi, jarak antara kedua mata menjadi jauh, tulang pipi menonjol.
Apakah pengobatan penyakit thalassemia?
Sampai saat ini belum ada obat yang menyembuhkan penyakit thalassemia secara total. Pengobatan yang paling optimal adalah transfusi darah seumur hidup dan mempertahankan kadar Hb selalu sama atau di atas 12 g/dl dan mengatasi akibat samping transfusi darah.
Apakah efek samping transfusi darah?
Efek samping transfusi darah adalah kelebihan zat besi dan terkena penyakit yang ditularkan melalui darah yang ditransfusikan. Setiap 250 ml darah yang ditransfusikan selalu membawa kira-kira 250 mg zat besi. Sedangkan kebutuhan normal manusia akan zat besi hanya 1-2 mg perhari. Pada penderita yang sudah sering mendapatkan transfusi kelebihan zat besi ini akan ditumpuk di jaringan-jaringan tubuh seperti hati, jantung, paru, otak, kulit dll. Penumpukan zat besi ini akan mengganggu fungsi organ tubuh tersebut dan bahkan dapat menyebabkan kematian akibat kegagalan fungsi jantung atau hati.
Bagaimana mengatasi kelebihan zat besi?
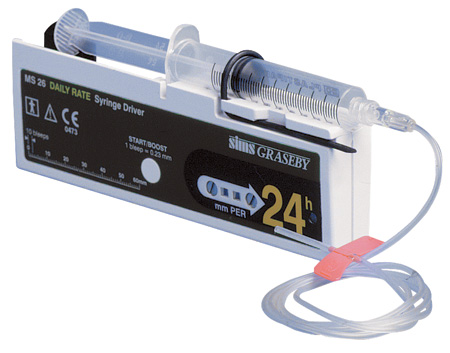
Pemberian obat kelasi besi atau pengikat zat besi (nama dagangnya Desferal) secara teratur dan terus menerus akan mengatasi masalah kelebihan zat besi. Obat kelasi besi (Desferal) yang saat ini tersedia di pasaran diberikan melalui jarum kecil ke bawah kulit (subkutan) dan obatnya dipompakan secara perlahan-lahan oleh alat yang disebut “syringe driver”. Pemakaian alat ini diperlukan karena kerja obat ini hanya efektif bila diberikan secara perlahan-lahan selama kurang lebih 10 jam per hari. Idealnya obat ini diberikan lima hari dalam seminggu seumur hidup.

Bagaimana mencegah kelahiran penderita thalassemia?
Kelahiran penderita thalassemia dapat dicegah dengan 2 cara. Pertama adalah mencegah perkawinan antara 2 orang pembawa sifat thalassemia. Kedua adalah memeriksa janin yang dikandung oleh pasangan pembawa sifat, dan menghentikan kehamilan bila janin dinyatakan sebagai penderita thalassemia (mendapat kedua gen thalassemia dari ayah clan ibunya).
Siapa yang harus diperiksa untuk kemungkinan pembawa sifat thalassemia?
Sebaiknya semua orang Indonesia dalam masa usia subur diperiksa kemungkinan membawa sifat thalassemia beta. Karena frekuensi pembawa sifat thalassemia beta di Indonesia berkisar antara 6-10%, artinya setiap 100 orang ada 6 sampai 10 orang pembawa sifat thalassimia beta. Terlebih lagi apabila ada riwayat seperti di bawah ini, pemeriksaan pembawa sifat thalassemia sangat dianjurkan:
Ada saudara sedarah yang menderita thalassemia beta.
Kadar hemoglobin relatif rendah antara 10-12 g/dl, walaupun sudah minum obat penambah darah seperti zat besi.
Ukuran sel darah merah lebih kecil dari normal walaupun keadaan Hb normal.
Ada saudara sedarah yang menderita thalassemia beta.
Kadar hemoglobin relatif rendah antara 10-12 g/dl, walaupun sudah minum obat penambah darah seperti zat besi.
Ukuran sel darah merah lebih kecil dari normal walaupun keadaan Hb normal.
Bagaimana prosedur diagnosis prenatal?
Diagnosis prenatal melalui beberapa tahap. Tahap pertama adalah pemeriksaan ibu janin yang meliputi pemeriksaan darah tepi lengkap dan analisis hemoglobin. Bila ibu dinyatakan pembawa sifat thalassemia beta maka pemeriksaan dilanjutkan ke tahap kedua yaitu suami diperiksa darah tepi lengkap dan analisis hemoglobin. Bila suami juga membawa sifat thalassemia maka suami-isteri ini diperiksa DNAnya untuk menentukan jenis kelainann pada gen globin beta.
Selanjutnya diambil jaringan janin (villi choriales atau jaringan ari-ari) pada saat janin berumur 10-12 minggu untuk diperiksa DNAnya. Bila janin ternyata hanya mebawa satu belah gen globin beta yang mengalami kelainan (gen thalassemia beta) atau sama sekali tidak membawa gen thalassemia beta maka kehamilan dapat diteruskan dengan aman. Tetapi bila janin ternyata membawa kedua belah gen thalassemia yang artinya janin akan menderita thalassemia beta maka penghentian kehamilan dapat menjadi pilihan.
Selanjutnya diambil jaringan janin (villi choriales atau jaringan ari-ari) pada saat janin berumur 10-12 minggu untuk diperiksa DNAnya. Bila janin ternyata hanya mebawa satu belah gen globin beta yang mengalami kelainan (gen thalassemia beta) atau sama sekali tidak membawa gen thalassemia beta maka kehamilan dapat diteruskan dengan aman. Tetapi bila janin ternyata membawa kedua belah gen thalassemia yang artinya janin akan menderita thalassemia beta maka penghentian kehamilan dapat menjadi pilihan.
Bagaimanakah prosedur dan apakah akibat tindakan pengambilan jaringan ari-ari terhadap janin?
Pengambilan jaringan janin dari ari-ari dilakukan dengan menusukkan jarum melalui jalan lahir atau dinding perut ke dalam alat kandungan clan menembus ke ari-ari, kemudian pada daerah ari-ari yang disebut villi choriales diambil dengan cara aspirasi sejumlah jaringan tersebut untuk bahan pemeriksaan DNA. Prosedur ini dilakukan oleh dokter ahli kandungan yang sudah berpengalaman melakukan tindakan ini. Prosedur ini dilakukan pada kehamilan 11 minggu. Tindakan ini mempunyai risiko keguguran sebesar 2-3%. Cara lain untuk mendapat sel dari janin adalah dengan pengambilan cairan amnion yang baru dapat dilakukan pada kehamilan 15 minggu. Risiko abortus pada prosedur ini adalah 1%.
Bagaimana mengetahui seseorang adalah pembawa sifat thalassemia beta?
Karena penampilan sebagian besar pembawa sifat thalassemia beta tidak dapat dibedakan dengan individu normal, maka pembawa sifat thalassemia beta hanya dapat ditentukan dengan pemeriksaan darah yang mencakup darah tepi lengkap clan analisis hemoglobin.
Dimana dapat diperiksa untuk kemungkinan pembawa sifat thalassemia?
Pemeriksaan pembawa sifat thalassemia beta dapat dilakukan di Lembaga Eijkman, Jalan Diponegoro 69 Jakarta pada setiap hari Senin s/d Jum’at jam 9.00 s/d 14.00. Biaya pemeriksaan adalah Rp. 150.000,-/per orang (harga sewaktu-waktu dapat berubah). Pemeriksaan ini juga dapat dilakukan oleh beberapa laboratorium lain.
Tiada ulasan:
Catat Ulasan